海洋生命・分子工学実験 II
(3) アガロースゲルからの DNA 断片の精製
はじ
めに
さて,次にしたいことは,制限酵素で
切断した DNA 断片を回収して mCherry cDNA 断片と
pBluescript II SK+ プラスミドベクターとを連結することです。しかし,mCherry cDNA を切り
出したときの pCR II 本体側の DNA 断片も,pBluescript II SK+ から切り捨てた EGFP cDNA
断片も (・・・どの断片も両端が BamHI と XhoI
の切断末端になっていますよね) 反応液に
一緒に入っています。これらを除去して,目的の DNA 断片のみを回収しないと,何を組み
立てているのかわからなくなります。
そこで,ここでは,まず制限酵素切断片をアガロースゲルで電気泳動して,それぞれの断片を
分離します。次に,目的の断片のみをゲルから切り出し,ゲルを溶かして DNA のみを精製
します。この DNA を,次の (4) で連結するわけです。
知っ
ておかなければならないこと
アガロースゲル電気泳動は「専門海洋生命・分子工学基礎実験」でやりました。憶えていますか?
アガロースは寒天の主成分です(図 1)。
図 1:
アガロースでできたゲル中に電場を作ると,マイナスに荷電した DNA はプラス極の方に向かって
移動します。アガロースの繊維は分子篩(ふるい)として働きます。短い DNA は篩の目に引っか
からないのでスルスルとすばやく移動し,長い DNA はあっちに引っかかりこっちに引っかかり
しながらゆっくりと移動するので, DNA を長さによって分離できるというわけです。長さのわかった
DNA 断片 (サイズマーカーという)を一緒に泳動すると,自分の調べたい
DNA 断片のおよその
長さがわかります(図 2)。
図 2: 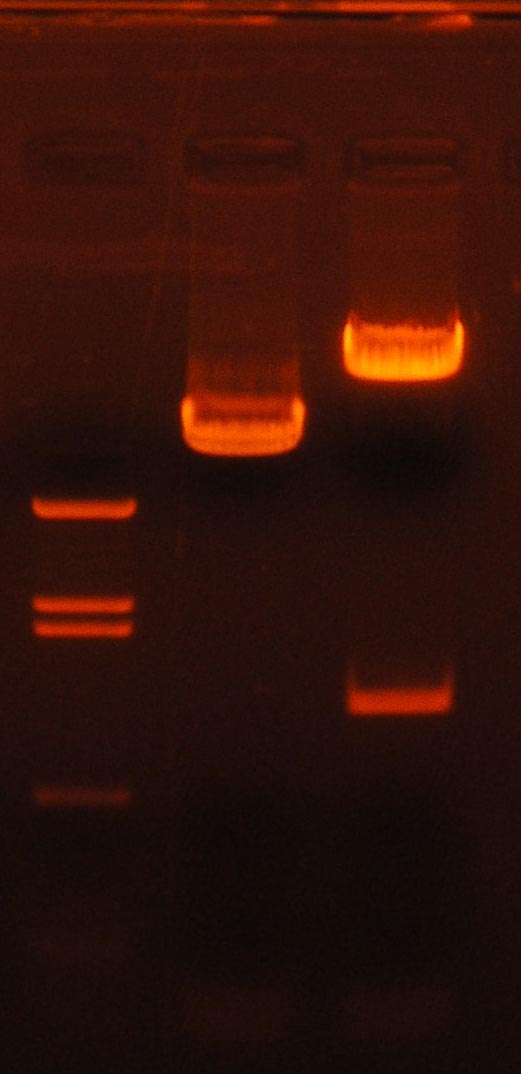
図 2 の電気泳動では,左端のレーンにサイズマーカーを泳動しまし
た。ここで用いたサイズ
マーカーはみんなが使うのと違う pBR322/BstNI です。サイズの大きいバンド (図の上側)
から順に(見えている分では) 1857 bp,1058 bp,929 bp,383 bp です。
今回の実習で、みんなが制限酵素
消化した DNA は、電気泳動でどのようなバンドに
なるか,もう予想はできていますか?
このような予想をすることは重要です。実際に電気泳動をしてみて,予想とちょっとでも違う
パターン(予想よりわずかに小さいとか,余分なバンドが出ているとか)になったとしたら,
以後の実験は大抵失敗します。それは,例えば制限酵素の Star 活性が出たためかも
知れ
ませんし,実験の計画そのものが間違っていた可能性を示唆するわけです。必ず,電気泳動の
前にどのようなサイズのバンドが
全部で何本出るはずかを予測しておいてください。
電気泳動の次には,目的のバンドを切り出して,そのゲル片から DNA を抽出・精製します。
これには市販のキット(タカラの
EasyTrap Ver. 2)
を使います。詳しくはキットの説明書を
配るので,それをよく読んでください。
実験
1.
エタ沈後の DNA
(10 μL TE に溶けている)に,6x 色素溶液 (a) を 2 μL 加える。
2.
1%
アガロースゲル (b) のサンプルコウムに全量を注ぎ込み,100 V で泳動する。
サイズマーカーとして λ/HindIII (c) を使います。
3.
トランスイルミネーターでバンド
を確認し,必要なバンドをゲルから切り出す。
カミソリでゲルを切り出す方法は,実験の当日実演して見せます。
切り出したゲル片は,新しいエッペンチューブに入れます。
4.
エッペンのゲル片を軽く遠心し
て,ゲルの大きさを推定する。
5.
ゲルの 3
倍量のヨウ化ナトリウム(NaI)溶液を加える。
6.
Easy Trap
ガラスパウダーを 5 μL 加え,ゲルが溶けるまで待つ。
注: ガラスパウダーは重いので固い沈殿になりやすいので,使用
直前によく
ボルテックスしてほぐしてから使いましょう。
注: ときどき指ではじいて混ぜ,ゲルが溶けるのを待ちます。
7.
ゲルが溶けたら室温で 5
分おく。その間ときどきボルテックスする。
ヨウ素イオンのようにイオン半径の大きい(chaotropic な)イオンの存在下で,DNA
がガラスビーズの表面に吸着します。
8.
3
秒ほど遠心し,上清を捨てる。
9.
洗浄用緩衝液を 500 μL
加え,ボルテックスで沈殿をほぐす。
10.
3
秒ほど遠心し,上清を捨てる。
11.
ステップ 9〜10
を,もう一度くり返す。
12.
TE を 20 μL
加え,ボルテックスで沈殿を完全にほぐす。
13.
55℃ で 2 分ほどおく。
注: 途中 1 回〜数回ボルテックスする。これで DNA
がガラスビーズからはがれる。
14.
5
秒ほど遠心し,上清を新しいエッペンに移す。
注: 今度は目的の DNA
断片が上清にあるので,間違って捨てないように!
15.
もう一度,とった上清を 5
秒ほど遠心し,新しいエッペンに上清を移す。
注: わずかの沈殿も取らないように細心の注意を払おう。
16.
上清の 5 μL
をアガロースゲル電気泳動でチェックする。
注: 回収した DNA
断片の量と純度を確認する。サイズも確認しよう。細くても,
はっきりとバンドが見えていれば,次のステップに進もう。
実験
に使う試薬
(a) 6x 色素溶液
この実験では [30% グリセリン,0.05% キシレンシアノール,0.05% ブロモフェノールブルー]
を使う。グリセリンは溶液を重くするため。ブロモフェノールブルーは泳動中にサンプルが
どの程度まで泳動されてきたかをモニターするための色素。1% アガロースゲルでは,
ブロモフェノールブルーは約 300 bp の DNA 断片とほぼ同じ位置を流れる。
(b) 1% アガロースゲル
1% のアガロース(
図 1)を
TAE
バッファー(d) に加熱溶解した後,臭化エチジウムを 0.5
μg/mL 加えて固めたもの。500 bp から 4 kb 程度のサイズの核酸を分離できる。臭化
エチジウムは発がん物質なのでゲルを素手で扱わないように。手についても問題はないが
洗った方が無難。
(c) λ/HindIII
λ ファージ を制限酵素 HindIII で切断した断片。各断片の長さは,移動度の小さい(泳動の起点に
近い)方から順に 23130 bp,9416 bp,6557 bp,4361 bp,2322 bp,2027 bp,564 bp,
125 bp である。ただし,1% 程度のアガロースゲルでは 564 bp のバンドはとても弱く,また
125 bp のバンドは見えない。またカプシド内から抽出精製した λ ファージ DNA は直線上で
両端には 10 nt 以上の付着末端(cohesive ends)を持つ。この部分は COS site と呼ばれる。
両端の付着末端どうしは相補的なので,うっかりするとそこが塩基対合してしまう。そこで,
λ/HindIII を扱う際には,常に氷中に起きうっかり温めないことが重要。実験台の上(室温)に
放置すると両端が結合してしまう。具体的には 23130 bp の断片と 4361 bp の断片が連結して
上から 4 本めのバンドがなくなり,一番上のバンドの位置が少しだけ上にずれる(こちらは
1% アガロースゲルでは気づけない程度のずれ)。
(d) TAE バッファー
40 mM Tris-acetate (トリスバッファーの pH を酢酸で約 8 に調整したもの)と 1 mM EDTA
を含むバッファー。通常, 50x 濃度のストック溶液を調製し,希釈して使う。
遺伝子工学的実験法の目次へ戻る