2‐9. 挿入 cDNA 断片の塩基配列決定
実験のデータはありません
はじめに
2-8 で行った実験では、できあがったプラスミド DNA を制限酵素で切断した
パターン(切れ方)を見ることによって、BvII ヘモグロビン cDNA が正しく
組み込まれているかどうかを判断しました。しかし、実際には BvII でない
DNA 断片が組み込まれているのに、偶然その制限酵素で同じような
切れ方をする場合がないとは言えません。より確実なチェック方法は
塩基配列の確認です。
また、この実験で PCR に用いた Taq DNA ポリメラーゼは他の多くの
DNA ポリメラーゼと比べて、誤ったヌクレオチドを取り込む確率が高い
(誤って取り込んだ部分を切り削って合成しなおす ”校正” 活性が弱い)
ため、組み込まれている配列が微妙に違っている可能性があります。
この実験では cDNA からタンパク質を発現させ、(実習ではやりませんが)
最終的には機能解析をすることを目的にしています。従って、たった 1 個
でもアミノ酸が違っていたりしたら結果の解釈がややこしくなってしまいます。
さらに、”稀に” ですが、制限酵素切断片を連結した箇所に思いもよらない
塩基の挿入や欠失がある場合があります。このような ”ずれ” が cDNA の
5' 末端側にあると、その場所からコドンの読み枠(フレーム)がずれてしまい
目的のタンパク質ができないことになります。
このようなことから、作ったプラスミドの塩基配列を決定しておくことは
非常に重要です。
知っておかなければならないこと
塩基配列の決定法として現在広く使われている方法は Sanger et al.
(1977) が
発明したジデオキシ法です。この方法の原理については他の講義で何度も
聴かされることと思います。ここでは原理については簡単に触れるだけにして、
より実際的な方法を説明します。実際には、実にさまざまなバリエーションがある
のですが、ここでは、この実習で実際に行う方法を中心にして解説します(図 1 参照)。
この方法では、塩基配列を決定したい DNA を鋳型として、それに相補的な
鎖を合成する反応を利用します。鋳型とする DNA は 1 本鎖でもいいし 2 本鎖の
DNA 断片(例えば PCR 産物)でもいいし、プラスミドそのままでも OK です。
塩基配列を決定したい DNA から RNA を合成して、それを鋳型とし、逆転写酵素で
相補的 DNA を合成する方法でも OK です。(今回の実験では 2 本鎖のプラスミド
DNA をそのまま鋳型として使います。)
いずれの場合も、基質となる dNTP に一定の割合でジデオキシリボヌクレオシド
三リン酸(ddNTP)を混ぜて合成反応を行います。 ddNTP には 3'
末端に
OH 基がないため、鎖の伸長がそこで止まってしまうという性質を利用しています
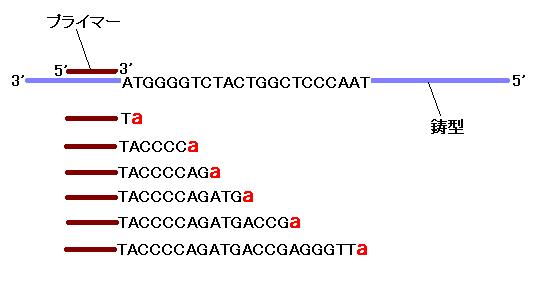
(図 1)。図 1 には反応液中に 4 種類の dNTP と ddATP を混ぜたときの反応
生成物を描いてあります。図では ddATP が取り込まれた部分を a で表し、
正常な dATP が取り込まれた箇所(A で表してある)と区別しています。どの程度
の割合で ddNTP を混ぜるかは使う酵素によって異なります。 ddNTP の割合が
高すぎると、プライマーに近い位置で全ての鎖の伸長が止まってしまい、
プライマーから遠い部分の配列が読めないなどの不都合が生じます。この ”割合”
は、使う DNA 合成酵素が ddNTP を ”どの程度の頻度で” 正常な dNTP と間違うか
によって決まります。
さて、このようにしてさまざまな箇所で合成が止まった、さまざまな長さの鎖を
”どうやって見るか” です。今回の実験では、ddNTP が蛍光色素で標識されて
います(蛍光色素が ddNTP に共有結合でくっついています)。プライマーを蛍光
標識する方法もありますし、昔は放射性同位元素で標識した dNTP で検出する
方法が一般的でした。今回の実験では、新たに合成されて ddNTP で伸長が
止まった鎖が、蛍光色素で標識されていることになるわけです。これをポリ
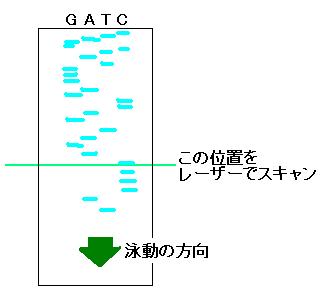
アクリルアミドゲル電気泳動によって分離します。今回用いる泳動装置では、
レーザーでゲルの一部分をスキャンして、短い順に流れてくる産物の蛍光を
検出し、パソコンに取り込む仕組みになっています(図 2)。
図 1
図 2
実験
前の実験で精製したプラスミド DNA を使う。1. dNTP/ddNTP 混合液 (a) をサンプル毎に 1 μl ずつ分注する。
dNTP/ddGTP、dNTP/ddATP、dNTP/ddTTP、dNTP/ddCTP を
別々に1 本ずつの PCR 用チューブに分注する。1 つのサンプル
(プラスミド)あたり 4 本のチューブが必要ということになる。
注: ddNTP が蛍光色素で標識されているので、強い光が当たらない
ようにアルミホイルなどで遮光した方がいいかも。
2. DNA/プライマー混合液を調製する。
以下のものを混ぜる
滅菌水を 19 μl
Miniprep DNA を 5 μl
反応バッファー (b) を 3.5 μl
プライマー (c) (1 pmol/μl)を 2 μl
ThermoSequenase (10 units/μl)を 2 μl注 1: 調製後は氷上におく。
注 2: チップを取りかえるなどして徹底してコンタミネーションを避ける。
注 3: 調製後、次のステップを行う前によく混ぜて遠心をする。
注 4: 失活を防ぐため、酵素は最後に加える。3. ステップ 2 で調製した混合液を、ステップ 1 で準備した各チューブ
に 7 μl ずつ分注する。4. ミネラルオイルをのせて、サーマルサイクラーで合成反応開始。
サイクルシークエンスの条件は以下のとおり。(1) [ 95℃ で 30 秒、60℃ で 20 秒、72℃ で 1 分 ]
を 30 回繰り返す。
(2) 4℃ で保温。注: アニーリングの条件はプライマーの種類によって変える必要がある。
5. エタノール沈殿で反応生成物を精製する。
各反応液をとり、パラフィルムの上をころがしてミネラルオイルを取り除く。
反応液を引きずると、ミネラルオイルがパラフィルムにくっつき、
水溶液(反応液)だけがチップについて来るはず。
ミネラルオイルを取り除けたら、以下の試薬を加える。
7.5 M 酢酸アンモニウムを 2 μl
エタノールを 30 μl6. 氷上に 20 分置いた後 20〜30 分遠心し、上清を捨てる。
7. 200 μl の 75% エタノールを加えて 5 分遠心後、上清を捨てて
沈殿を乾燥させる。8. 6 μl の loading dye (d) を加える。
9. 70℃ で 3 分加熱した後、氷で急冷する。
10.2 μl ずつ電気泳動ゲルにアプライする。
注: 1 サンプルをアプライしたら、一度カバーを閉じて 20 秒泳動を
行い、サンプルを沈める。その後、次のサンプルをアプライする。* 電気泳動装置、データ解析の方法については説明を省略します。
説明書があるので、見たい人は言ってください。
実験に使う試薬
各 150 mM の dNTP と、 3 mM の ddGTP、ddATP、ddCTP、ddTTP いずれか(b) 反応バッファー
の混合液。ddNTP は Cy5.5 という蛍光色素で標識されている。
150 mM Tris-HCl (pH 9.5)、 35 mM MgCl2(c) プライマー
今回使うプライマーは組み込んだ cDNA の上流(オペレーター部分)に(d) Loading dye
対するプライマーで(2-10、2-11 参照)、配列は以下のとおり。
[5'-TTTGTGAGCGGATAACAATTA-3']
ホルムアミドと色素を含んでいますが、組成はわからない(秘密?)らしい。
ホルムアミドは DNA を変性させ(2 次構造ができないようにし)、電気泳動の
ときに、DNA の塩基長に応じた正確な分離を可能にする。色素を加えるのは
サンプルをアプライしたときに(隣のレーンに漏れたりしていないかどうか)
目で見て確認するため。