2‐4. PCR 産物とプラスミドベクターの制限酵素処理
はじめに
PCR 産物は、そのままでは”使ったらなくなってしまいます。”そこで
これをプラスミドに組み込んで、大腸菌に導入します。この一連の
作業を”クローニング”といいます。プラスミドは大腸菌の中で
大腸菌の染色体(本来染色体というべきではないのですが・・・)
DNA と独立に複製して増殖する遺伝因子です(「分子遺伝学C」で
習ったはず)。一旦、cDNA がクローニングされれば、プラスミド
を導入した大腸菌を培養して cDNA を無限に増やすことができるし、
大腸菌を利用してタンパクを発現させることも可能です。
cDNA や遺伝子 DNA のクローニングには、プラスミド以外にも
バクテリオファージなどを使うことができます。cDNA や遺伝子を
組み込むためにデザインされたプラスミドやファージを総称して
ベクターといいます。また宿主としても、大腸菌のみでなく、酵母や
昆虫、哺乳類など真核生物の細胞も利用できます。
ここでは、増幅したヘモグロビン cDNA をプラスミドベクターに
組み込むために、PCR 産物の両末端の形を整えます。そのために
使うのが制限酵素です。制限酵素は特定の配列をした 2 本鎖
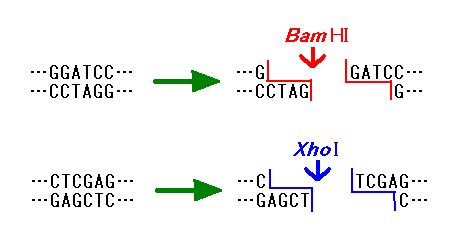
DNA を認識してそれを切断する酵素の総称です。例えば、BamHI
という制限酵素は [GGATCC] という配列を認識して、これを
[G / GATCC] という風に切断します(図 1)。
図 1 BamHI と XhoI という 2 つの制限酵素が DNA を切断する様式。
切断端の形状が同じ DNA 断片どうしは、再び連結することができます。
このとき用いるのは、DNA の修復に関わる酵素(DNA リガーゼと
いいます・・・これについては 2-6 で説明します)です。これらの酵素類の
おかげで、私たちは
遺伝子や cDNA を自由自在に切ったり貼ったりできる
のです。これが遺伝子工学の真髄です。こういった作業の原理や方法
は非常にシンプルであり、しかもほとんど全ての生物に応用できます。
そして何よりも、遺伝や遺伝子の本質に直接迫るこういった実験法の
確立は、生命科学の爆発的な進歩を可能にしたのです。
知っておかなければならないこと
さて、みなさんが増幅したヘモグロビンの cDNA はどんなプライマーで増幅
したものでしょうか?
ヘモグロビン特異的プライマーは
[5'-AAAGGATCCATGAGTAAACCAGCTGAAGCC-3']
アダプタープライマーは
[5'-GACTCGAGTCGACATCG-3']
です。何か気づきましたか?(気づくよな)
ヘモグロビンプライマーの方には BamHI 切断部位が、アダプタープライマーの
方には XhoI 切断部位があります(それぞれ赤字で示してあります)。もう一つ
大切なことですが、2-3 のところに載せたヘモグロビン cDNA の塩基配列には、
どこを探しても BamHI や XhoI の切断部位はありません。つまり、皆さんが
増幅した cDNA を BamHI と XhoI の両方で切断すると、cDNA の 5' 末端には
BamHI の切断端が、3' 末端には XhoI の切断端ができます。
これをプラスミドに組み込みたいのですから、もともと環状であるプラスミド DNA
の方も切断して、片方の末端を BamHI の、もう片方の末端を XhoI の形に
したいですね。今回使うのは、 QIAGEN 社の pQE-30 というプラスミドです。
このプラスミドは以下のような構造をしています(図 2)。
このプラスミドには XhoI 切断部位が1ヶ所ありますが、ここは使えません。
(なぜ使えないかはこのページと 2-9 から読み取ってください。)
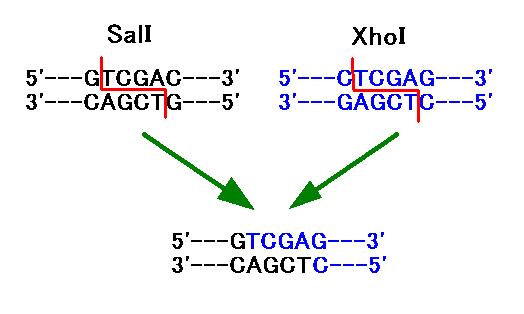
そこで、プラスミドの方は、SalI で切断します。SalIの認識配列は
[GTCGAC] です。SalI の切断片と XhoI
の切断部位は、同じ形をしている
ので、そのまま連結することができるのです(図 3)。要するに、図 2 の
プラスミドから、 BamHI と SalI にはさまれた短い DNA 断片を切り捨て、
ここに BamHI と XhoI で切断した PCR 産物を挟み込むわけです。
図 2 プラスミドには大腸菌内で複製するための複製開始点(ori)が
あります。また、ampr 遺伝子(アンピシリン耐性遺伝子)は
β‐ラクタマーゼという酵素をコードしています。この酵素は、ペニシリンや
アンピシリンなどのようにラクタム環という
4 員環を持つ抗生物質を
分解します。大腸菌は抗生物質存在下では生育できませんが、プラスミドを
導入された大腸菌は抗生物質入りの培地で生育できます。したがって、
プラスミドを大腸菌に導入した後抗生物質入りの培地にまけば、プラスミドを
持つ大腸菌だけを選択的に増やすことができるという仕組みになっています。
それ以外の部分については、2-9 のページで詳しく説明します。
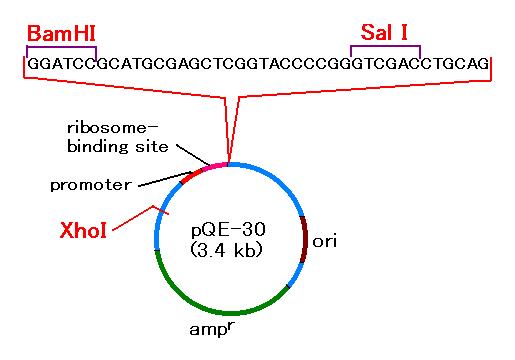
図 3 SalI 切断部位と XhoI
切断部位は連結できる。
ただし、このような連結をした後は、この位置を SalI で切ることも
XhoI で切ることもできなくなる。
実験
[ PCR 産物の制限酵素処理 ]
注: ステップ 1〜8 は時間の都合で省略するかも。その場合は、[ プラスミドベクターの制限酵素処理 ]
PCR の反応液を 10 μl とって、ステップ 9 へ進む。1. PCR の反応液を新しいエッペンに移す。
注: ミネラルオイルをあまりたくさん取らないように。
2. 等量以上のフェノール/クロロホルム(a) を加えボルテックス。3. 遠心後、上層を新しいエッペンに移し、その 10 分の 1 量の
3 M 酢酸ナトリウム(b) を加え、よく混ぜる。4. 上記の水溶液の 2.5 倍量のエタノールを加えてよく混ぜ、遠心する。
エッペンのフタの蝶番を外側に向けてローターにセットする。
エッペンは斜めの角度を保ったまま回転するので、沈殿は
一番底ではなくて、底に斜めにくっつくように沈む。5. 沈殿を取らないようにエタノールをできるだけ完全に捨てる。
6. 75% エタノールを 100 μl 程度加え、再び数秒遠心する。
7. 沈殿を取らないようにエタノールをできるだけ完全に捨てる。
8. 沈殿を乾燥させ、滅菌水を 50 μl 加えて溶かす。
9. このうち 10 μl を新しいエッペンにとり、以下の試薬を加える。
滅菌水を 12 μl10. よく混ぜた後、37℃ のウォーターバスに 1 時間おく。
10x 制限酵素バッファー2(c) を 3 μl
[100 mM Tris-HCl (pH 7.9), 500 mM NaCl, 100 mM MgCl2,
10 mM DTT]
1 mg/ml ウシ血清アルブミンを 3 μl
BamHI を 1 μl
XhoI を 1 μl
1. 新しいエッペンに、滅菌水を 19 μl とる。[ 制限酵素切断片の精製と、アガロースゲル電気泳動 ]2. 以下の試薬を加え、よく混ぜる。
プラスミドベクター pQE-30 (約 1 μg) を 3 μl3. 37℃ のウォーターバスに 1 時間おく。
10x 制限酵素バッファー(BamHI 用)(c) を 3 μl
[100 mM Tris-HCl (pH 7.9), 1.5 M NaCl, 100 mM MgCl2,
10 mM DTT]
1 mg/ml ウシ血清アルブミンを 3 μl
BamHI を 1 μl
SalI を 1 μl
制限酵素で切断した PCR 産物とプラスミドベクターは、それぞれ、
切り捨てるべき小さな DNA 断片を除去して精製しなければいけない。
そうしないと、連結反応をしたときに元通りの組み合わせで連結
してしまい、PCR 産物とプラスミドとの連結の効率が著しく悪くなる
からである。
そのため、制限酵素処理後の DNA を電気泳動し、目的の断片
だけをゲルから切り出して精製する。これによって、PCR 産物の
両端から切り落とされる数塩基の断片と、プラスミド(図 2 参照)
の BamHI-SalI 間の短い断片も除去することができる。
ここでは、とりあえず電気泳動を行ってゲルからバンドを切り出して
おき、次回にそのゲル片から DNA を精製する。1. 制限酵素処理後の PCR 産物とプラスミド DNA に
6x 色素溶液(d) を 5 μl 加えて、1% アガロースゲル(e) で
電気泳動をする。
サイズマーカーには φX174/HaeIII(g) を 7.5 μl 使う。
注: PCR 産物とプラスミドと、マーカーのバンドとの位置関係を
きちんとイメージしておくこと(図 4)。2. PCR 産物とプラスミドのバンドをトランスイルミネーターで
確認し、ゲルからバンドを切り出す。
切り出したバンドはそのままエッペンチューブに入れて、
次回まで -20℃ で冷凍保存しておく。
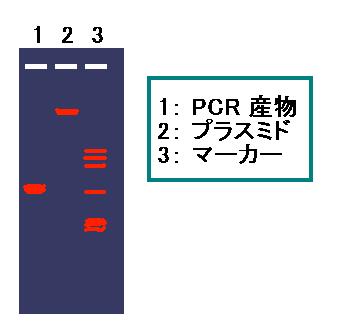 図 4
図 4
実験に使う試薬
水あるいは Tris-HCl バッファーで飽和したフェノール(2-1 参照)(b) 3 M 酢酸ナトリウム
とクロロホルムを等量混ぜたもの。消泡剤としてイソアミルアルコール
(3-メチル-1-ブタノール)を少量加えることもある。
酢酸ナトリウムの粉を最終濃度 3 M になるように溶かし、酢酸で pH を(c) 10x 制限酵素バッファー
5.2 に合わせたもの。核酸のエタノール沈殿用の塩としては最も一般的
に使われる。エタノール沈殿のときは 0.3 M になるように核酸溶液に
加え、さらに 2-2.5 倍量のエタノールを加えて核酸を沈殿させる。
制限酵素には非常にたくさんの種類があるが、それぞれに最適条件(d) 6x 色素溶液
が違う。特に塩濃度は重要で、市販の制限酵素には、それぞれに
最適な塩濃度のバッファーが添付されている。BamHI と XhoI は
同じ条件で使えるので、今回は同時に処理する。最適条件の異なる
2 つの制限酵素を使いたいときは、1 度片方の制限酵素で切断して
から、フェノール/クロロホルム抽出、エタノール沈殿をして、もう一方の
制限酵素で切断しなければならない。
また、制限酵素の活性は他のいろいろな条件によって影響を
受ける。例えば、酵素は、安定に保たれるために 50% のグリセリン
の入った状態で保存されているが、酵素反応液中のグリセリン濃度が
5% を超えると、本来切らない配列を切ってしまう(認識配列が甘くなる)
現象が見られる。例えば、BamHI の認識配列は [GGATCC] であるが
これが [GGATCT] といったような、”似てるけど違う”配列を切ることが
ある。このような活性のことを Star 活性という。グリセリン濃度を低く
抑えるためには、酵素を反応液全体の量の 10 分の 1 以上には
しないなどの注意が必要である。
この実験では、[30% グリセリン、0.05% ブロモフェノールブルー](e) 1% アガロースゲル
を使う。グリセリンは溶液を重くするため。ブロモフェノールブルーは
泳動中にサンプルがどの程度まで泳動されてきたかをモニター
するための色素。1% アガロースゲルでは、ブロモフェノールブルー
は約 300 bp の DNA 断片とほぼ同じ位置を流れる。
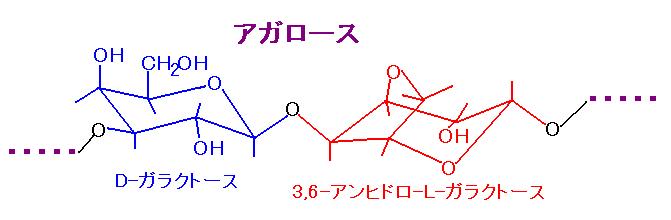
1% のアガロース(アガロースは寒天の主成分・・・図 5)を、図 5
TAE バッファー(f) に加熱溶解した後、臭化エチジウムを 0.5 μg/ml
加えて、固めたもの。500 bp から 4 kb 程度のサイズの核酸を
分離できる。臭化エチジウムは発がん物質なのでゲルを
素手で扱わないように。手についても問題はないが、洗った方が無難。
40 mM Tris-acetate (トリスバッファーの pH を酢酸で約 8 に調整(g) φX174/HaeIII
したもの)と 1 mM EDTA を含むバッファー。通常、50x 濃度のストック
溶液を調製し、希釈して使う。
φX174 という小さなバクテリオファージの DNA を HaeIII (認識配列は
[GGCC]) という制限酵素で切断したもの、大きい方から順に、
1353 bp、1078 bp、872 bp、603 bp、310 bp、281 bp、
271 bp、234 bp、194 bp、118 bp、72 bpの断片があるが、
1% アガロースゲル電気泳動では 310 bp より小さいバンドはきれいに
分離しない。