6. アガロースゲル電気泳動と
ゲルからの DNA の精製
《 みんなのデータ 》
はじめに
次にしたいことは、制限酵素で切断した cDNA 断片と pQE30
プラスミドとの連結です。
ここで、注意すべきことは、制限酵素消化の反応液中には、自分のほしい cDNA と
pQE30 プラスミドだけではなく、pBluescript II SK+
プラスミドの断片などといった
不要な DNA 断片も存在することです。しかも、それらの断片の末端の形状も、当然の
ことながら BamHI や HindIII によって作られた付着末端です。したがって、連結反応を
させる前に不要な DNA 断片を除去したいですね。ここで行うのは、制限酵素切断片の
アガロースゲル電気泳動と、ゲルからの DNA 断片の精製です。
知っておかなければならないこと
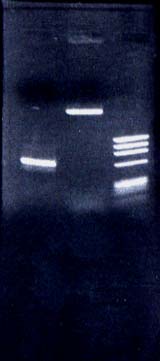
実験
1. 制限酵素処理後の DNA (10 μL の TE に溶けている)に 6x 色素溶液(a)
2. トランスイルミネーターでバンドを確認し、必要なバンドをゲルから切り出す。
を 2 μL 加えて、1% アガロースゲル(b) で 電気泳動をする。サイズマーカー
には λ/HindIII(d) を 10 μL 使う。
新しいエッペンを用意し、切り出したバンドを入れる。
3. エッペンのゲル片を軽く遠心して、ゲルの大きさを推定する。
4. ゲルの 3 倍量の NaI (ヨウ化ナトリウム)溶液を加える。
ゲル片が約 100 μL なら NaI 溶液を約 300 μL 。5. 55℃ でゲルを溶かした後、室温に戻す。
6. Easy Trap ガラスパウダーを 5 μL 加え、よく混ぜる。
ガラスパウダーは硬い沈殿になっているので、使う前に念入りにボルテックス
して、ほぐしておく。7. 室温で 5 分間おく。
ときどきボルテックスをして、ガラスビーズが沈まないようにする。ヨウ素イオン
のようにイオン半径の大きい(chaotropic な)イオンの存在下で、DNA がガラス
ビーズの表面に吸着する。8. 3 秒ほど遠心し、上清を捨てる。
9. 洗浄用緩衝液を 500 μL 加え、ボルテックスで沈殿をほぐす。
10. 3 秒ほど遠心し、上清を捨てる。
11. ステップ 7 とステップ 8 を繰り返す。
12. 沈殿に TE バッファーを 20 μL 加え、ボルテックス。
沈殿を完全にほぐす。13. 55℃ で 2 分ほどおく。
ときどきボルテックス。・・・これで、DNA がガラスからはがれる。14. 5 秒ほど遠心し、上清を新しいエッペンに移す。
沈殿を取らないように。15. もう 1 度 5 秒ほど遠心し、上清を新しいエッペンに移す。
16. 上清の 5 μL 分をアガロースゲル電気泳動でチェック。
絶対に沈殿を取らないように。
精製された DNA 断片の量と純度を推定する。
バンドとして見える程度の量があれば、次のステップに進める。
実験に使う試薬・器具
この実験では、[30% グリセリン、0.05% キシレンシアノール、0.05% ブロモフェノール(b) 1% アガロースゲル
ブルー] を使う。グリセリンは溶液を重くするため。ブロモフェノールブルーは泳動中
にサンプルがどの程度まで泳動されてきたかをモニターするための色素。1%
アガロースゲルでは、ブロモフェノールブルーは約 300 bp の DNA 断片とほぼ同じ
位置を流れる。
1% のアガロース(アガロースは寒天の主成分・・・図 5)を、 TAE バッファー(c) に(c) TAE バッファー
加熱溶解した後、臭化エチジウムを 0.5 μg/mL 加えて、固めたもの。500 bp から
4 kb 程度のサイズの核酸を分離できる。臭化エチジウムは発がん物質なので
ゲルを素手で扱わないように。手についても問題はないが、洗った方が無難。
40 mM Tris-acetate (トリスバッファーの pH を酢酸で約 8 に調整したもの)と(d) λ/HindIII
1 mM EDTA を含むバッファー。通常、50x 濃度のストック溶液を調製し、希釈して
使う。
野生型の λ ファージを HindIII で切断した断片。大きい方から 23.13 kb、
9.42 kb、6.56 kb、4.36 kb、2.32 kb、2.02 kb、0.56 kb、0.13 kb。上述の
φX174/HaeIII マーカーよりも比較的大きな断片が多い。2 つのマーカーを
あわせて使う人が多い。λファージを制限酵素で切断したマーカーは他にも
たくさんあるが、注意することがある。λファージはファージ粒子の内部では
直鎖状であり、大腸菌に感染すると環状になる。実はファージのゲノム DNA
の両端は相補的な付着末端(cos site という)になっており、この部分が
ファージが閉環状になるときの両末端の連結に重要なのである。また、ファージ
が大腸菌内で新たな殻の中に入るときには直鎖状のファージゲノム DNA が
タンデムに多数連結したもの(concatemer という)が生成する。このときにも
直鎖状のファージ DNA どうしは cos site で連結している。λファージ由来の
サイズマーカーを使うときには、凍結保存してあるマーカーを溶かした後
すぐに氷につけておき、決して温めてはいけない。温めると、23.13 kb のバンド
と 4.36 kb のバンドがくっついてしまう。