生体機能物質工学実験 II
第 3 週 + 第 4 週のレポートへの簡単なコメント
講評:
今回のレポートは、1 回めのレポートが返ってきてから後に提出したものですよね?
1 回めのレポートで指摘されたことと同じことを指摘されている人はいませんか?
コメントをきちんと読んで、次の回に活かしていますか?
レポートの中に、他人の書いた文章をコピー&ペーストしただけのものが見られます。
最初から最後まで同一でなく、自分なりに加筆修正していればそれはいいでしょう。ただ
(たとえ一部分であっても)完全に同じ部分があるというのは、レポートを読む側にとっては
おもしろくありません。一生懸命考えてレポートを書いていると思うから、隅々までよく読んで
少しでも役に立つコメント・アドバイスをしようと思うのです。努力せずに書いたレポートは
読む気がしません。大学は義務教育ではないから、あなたたちは勉強する義務はありません。
したくない人はしなくてよいです。そのかわり勉強したくない人に教えることは何もありません。
学ぶ意欲のない人のために使う時間は無駄です。その分、意欲のある人のために使うのが
当然です。私は、「できる」ことを要求しているのではありません(それは実習のときにも
何度も言ったはず)。「考える」ことが大切なのです。たとえ、正解に辿り着けなくても、そんな
ことは問題ではないのです。他の人と比べる必要もありません。ただ、自分のできるだけの
ことをしてくれればそれでいいのです。どう?
課題 1:
* 実験の「方法」の書き方は、よくなりました(全員ではないけれども・・・)。ただ、「結果」が
方法のところに付け足しのように簡単に書いてあるだけの人が多いです。しっかり、
「結果」を記述して、その結果について考察してください。
* 解析した塩基配列を、きちんと「結果」として書いている人が案外少なくて残念です。予想
通りの箇所に変異が導入されていない(元どおりの)配列がたくさんありました。また、
予想外の場所に変異の入ったクローンもありましたね。これらの原因は、前回のレポート
課題でいろいろと考えましたが、早速それを実際に体験することができましたね。それ
なのに、考察をしていない人がいます。ただ言われたとおりに作業をするだけでは実習の
目的・目標は半分も達成されません。自分のやった実験の内容、結果についてよ〜く考え
ないと、何も身につきませんよ。来年、卒業研究で、同じ実験方法を一から教わらなくても
いいように、しっかり自分のものにしてくださいね。
配列が期待通りであったかどうかは、重要な問題ではありません。期待はずれの結果
を得たことは “チャンス” です。その原因を考えることはとても重要なことです。
* 制限酵素による切断が不完全なサンプルが多かったと思います。これについての考察を
している人もあまり多くありませんでした。ここをしっかり考えておくと、課題 3 でも何か
思いついたかもしれません。。。
今回は実習ということで仕方なく強行しましたが、もしも普通の実験でこのような結果が
出たら、あらためて、完全に切れるような条件で(プラスミドを減らしたり、酵素の濃度を
上げたり、時間を長くしたり・・・)制限酵素処理をするところからやり直します。それが
一番です。
* エタノール沈殿のところで、「エタノールを加えることにより水を蒸発させる」と書いてある
人が結構たくさんいましたが、エタノール沈殿でエタノールを加えるのは核酸の水和水を
奪って沈殿させるのが目的です。
課題 2:
* 課題 2 は簡単でしたね。わからなかった人、わかった人に教えてもらってください。
課題 3:
* PCR で増幅した cDNA の両端付近に BamHI 配列と HindIII 配列があります。でも PCR の
反応生成物の両末端は制限酵素の付着末端になるはずもありません。だから、両端を
制限酵素の付着末端にしてやらないといけません。ところが cDNA の内部に HindIII 配列
があるので、HindIII 処理によって末端の形状を整えてやろうとすると内部でも切れてしまう。
この問題をどうやって解決するか、というのが課題です。
* 「内部の HindIII がなくなればいい」 ・・・ストレートな発想でよいですね。今回の実習では
部位特異的突然変異を学んだので、同じ方法で・・・。というのは・・・どうでしょう? Kunkel
法で変異を入れるためには cDNA を pBluescript のようなプラスミドに組み込まないと
いけない。でも、組み込むために変異を入れないといけない。これでは堂々巡りです。
プラスミドに組み込まずに cDNA に変異を入れる方法はあります。詳しくは宮内さんに教え
てもらってください。
* 「内部の HindIII で切れなければいい」 ・・・これも同様にストレートな発想です。具体的に
はどうするかというと、計 2 ヶ所ある HindIII 配列のうち 1 ヶ所しか切れないような部分分解
(部分消化)をすればいいのです。数十キロベースを超えるようなゲノム
DNA 断片をプラス
ミドにクローニングしようとする場合に、こういう方法をとる人がいます。具体的には、HindIII
の濃度を低くする(10 分の 1 とか 100 分の 1 とか・・・何段階か試してみないといけません)
そして反応時間を短くする(5 分とか 10 分とか)などの方法がとられます。みんな、実際、
今回の実習ではプラスミドを完全に切ろうと思ったのに、切れるはずの配列が切れていない
ような断片がたくさん見られましたよね。それがヒントにもなったはずです。
この方法は比較的ポピュラーです。ただし、2 ヶ所の HindIII の配列を区別することはでき
ませんから、内部の配列が切れる場合、末端付近の配列が切れる場合、どちらも切れる
場合、どちらも切れない場合の全ての切れ方が起こります。でも、これらの断片の混合物の
中から、目的の断片を電気泳動で区別し回収することができれば実験は成立しますよね。
今回の課題の場合、末端付近の HindIII 配列が切れた場合と切れなかった場合とで、
cDNA のサイズがほとんど違わないので、これらを区別するのは難しいだろうと思います。
* 私がよくやる方法は、以下のような方法です。まず cDNA (PCR 産物)の一部を BamHI と
SacI で切断します(図 1A)。それと別の一部を SacI と HindIII
で切断します(図 1B)。
図 1 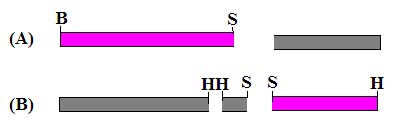
もちろん、プラスミドの方は BamHI と HindIII で切断しておきます。この場合は部分分解では
なく、完全に切るようにします。そして 3 つのサンプルをそれぞれ別々に(3 レーン使って)
電気泳動します。このとき、上の図 1A と図 1B で生じるピンクの断片を回収します。それぞれ
長さが違うので、電気泳動で区別できますよね。もしも不要な断片のうちどれかが自分の
ほしい断片とほとんど同じ長さである場合には、不要な断片の真ん中あたりを切るような
(しかしそれ以外の断片を切らないような制限酵素も同時に働かせて不要な断片をさらに
細かく切る、というようなことも考えます)。これらの断片をプラスミド断片とライゲーション
させます(図 2)。
図 2 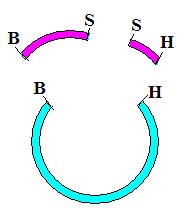
この場合、各連結部分における末端の形状はそれぞれ違いますよね。だから、狙いと
違うパターンで断片が連結してコロニーとして回収されることはありません。 2 つの断片
をライゲーションするのと比べて多少効率は落ちると思いますが、これまで何度もこう
やってプラスミドを作ってきた経験から言うと、それはあまり問題にはなりません。この
方法だと、EasyTrap を用いて、電気泳動ゲルから DNA 断片を回収する作業は 1 回で
済みますよね?
* 強引に BamHI と HindIII で分解して 3 つの断片でライゲーションをしようというアイデアは
あまり現実的ではありません。
図 3 
このような方法だと、BamHI⇔HindIII 断片の HindIII 末端もプラスミドとつながることが可能
ですよね。こういう場合には、圧倒的多数のコロニーが BamHI⇔HindIII 断片のみを組み
込んだプラスミドを持つことになると予想されます。なぜかは、考えてみてください。また、
可能性としては、HindIII⇔HindIII 断片が挟まるとしてもどちら向きに挟まるか、あるいは
いくつ挟まるかも不確定となりいろいろな可能性のある中から正しいつながり方をした
プラスミドを選ばないといけません。たくさんのコロニーを拾っても 1 個も本物がないという
結果になるのが落ちです。
* BamHI と SacI と HindIII で同時に切断して(図 1B のようになりますよね・・・)、これらを
ライゲーションさせる。ただし、まず図 1B のピンクの断片(SacI⇔HindIII)とプラスミドを
ライゲーションさせる(断片 X としましょうか)。それと別に図 1B のグレーの断片どうし
をライゲーションさせる(これを断片 Y としましょう)。その後、断片 X と断片 Y とを
ライゲーションさせるというアイデアがありました。こういう手順を踏めば、各ステップで
可能な連結の仕方は 1 とおりしかないので、期待通りのプラスミドが組み立てられる・・・。
・・・本当にそうかな???
結構たくさんの人がこう考えましたが、この方法にはいくつかの落とし穴があります。
まず、断片 X のライゲーションのとき、本当に可能な組み合わせが 1 とおりしかない
でしょうか?たとえばピンクの断片どうしだって HindIII 末端どうしで連結できますよね?
断片 Y の場合も、例えば BamHI⇔HindIII 断片どうしで連結できますね。そのような
期待はずれの形をした断片どうしが、第 2 段階のライゲーションでさらに結合して大混乱
になってしまいます。ただ、このような “期待はずれ” の連結は、例えば今回の実習の
ような単純なライゲーションの場合にも起こり得ます(もっとずっとシンプルではありますが)。
ただし、cDNA どうしが連結した場合には複製可能なプラスミドはできないし、プラスミドが
2 個、BamHI と HindIII 末端どうしで結合する確率は非常に低いです(短いものの方が
連結しやすいのか・・・実際そういうクローンを拾った経験がありません)。
それよりも大きな問題は 2 段階のライゲーションです。前回のレポートで散々悩まされた
ことを思い出し、活かしてください。 In vitro の反応では 100% ということはまず起こりま
せん。ライゲーション反応では、連結されずに残る断片が必ずできます。これを取り除かず
に次のステップに行くとどうなるか・・・。例えばこういうことが起こります。断片 X を作る
ライゲーション反応が起こらなかったプラスミドは元どおりに BamHI と HindIII 末端を持って
います。一方、断片 Y を作る際にも、反応しなかった BamHI⇔HindIII 断片が残ります。
断片 X と断片 Y をライゲーションさせているつもりが、上記の未反応の断片どうしの
ライゲーションが起こって、結局一番左側の BamHI⇔HindIII のみが組み込まれたプラスミド
ばかりが取れてくることになります。“それなら” ということで断片 X と断片 Y をライゲー
ションさせた後で、結合した分だけを電気泳動で精製しようということになれば、結果として
これが 2 回めの電気泳動となり、おそらくほとんどの断片は回収できずに失われていく
ことになります。よく考えてあったけれど、残念ながらうまく行きそうにありません。
* HindIII リンカーをつけるという方法を考えた人もいました。どうするかというと図 4 のように
HindIII 付着末端を持つ合成オリゴヌクレオチドを、PCR 産物の両末端にライゲーション
させるのです。
図 4 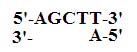
実際には、二本鎖部分をもう少し長くしておかないといけません。PCR 産物を BamHI で
切断すると、BamHI 側の末端に連結されたリンカーは BamHI による切断で切り落とされて
しまいます。ここでは HindIII を使いませんので、内部の HindIII 部位を切ることなく、末端
に HindIII 付着末端を作ることができます。リンカーのライゲーションの場合、合成オリゴを
多量に入れて反応をするので、電気泳動によって DNA を精製する必要はないので、実現
可能かと思います。cDNA を単離するときにはこれと似た方法でクローニングするような
プロトコルもあります。
実験のもくじのページに戻る