5. pBluescript II SK+ ベクターからの
EGFP cDNA の切り出し
《 みんなのデータ 》
はじめに
さて、期待したとおりの突然変異の導入された cDNA はできていましたか?塩基配列を
確認したら、今度は変異の入った cDNA から変異型タンパクを発現させたいですね。
第 1 章のページをもう一度見てください。変異導入の作業をするときには cDNA を
pBluescript II SK+ というプラスミドに組み込んであります。憶えていますか?
・・・そんなわけで、変異を導入した cDNA をタンパク発現用のプラスミド(pQE30)に
移し替える作業をしなければいけません。ここでは、まず、制限酵素を利用して、
cDNA を切り出します。
知っておかなければならないこと
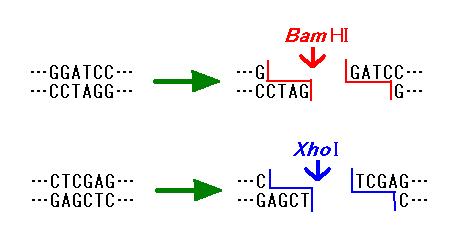
図 1
切断端の形状が同じ DNA 断片どうしは、再び連結することができます。同じ制限酵素で
切断した末端どうしは当然再連結させることができます。また、平滑末端どうしなら
どんな制限酵素で切断した末端どうしでも連結できます。さらに、異なる配列を認識する
制限酵素で切断した末端どうしであっても、突出部分の配列がたまたま同じであれば
連結することができます(図 2)。図 2 の例では、SalI
と XhoI で切断した末端どうし
を連結させています。できあがった配列に注目してください。[5'-GTCGAG-3']
という
この配列は SalI と XhoI どちらの認識配列とも違います。従ってこのような再連結を
した後の配列は、もう SalI でも XhoI でも切断することはできません。
図 2 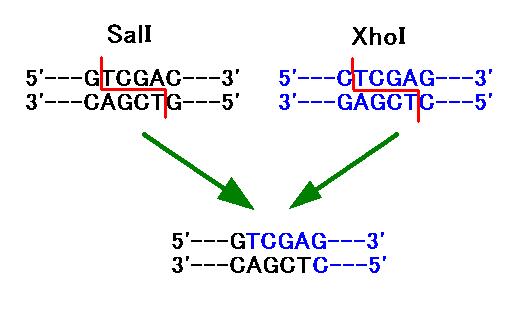
DNA の末端どうしの連結に使うのは、DNA の修復や複製に関わる酵素(DNA
リガーゼと
いいます・・・「分子遺伝学 C」の講義を憶えているかな?)。これらの酵素類の おかげで、
私たちは遺伝子や cDNA を自由自在に切ったり貼ったりできるのです。
これが遺伝子工学の真髄です。こういった作業の原理や方法は非常にシンプルであり
しかもほとんど全ての生物に応用できます。そして何よりも、遺伝や遺伝子の本質に
直接迫るこういった実験法の確立は、生命科学の爆発的な進歩を可能にしたのです。
制限酵素を利用するときに、注意すべき点は、反応液の組成です。酵素の種類によって
反応に最適な塩濃度や pH などが違います。塩濃度が高すぎたり低すぎたり、また
pH が大きくずれていたり、また酵素の濃度が高すぎたりすると、どういうことが起こるか
というと、本来なら切らないはずの(認識配列と似ているけれども完全に同じではない)
配列を切ってしまうのです。これを制限酵素の Star 活性といいます。今回の実習でも
使う BamHI は非常に Star 活性の出やすい酵素です。例えば、本来の認識配列
[GGATCC] と似ている [GGATCT] などといった配列を切る可能性があります。
実習では、反応液の組成がでたらめにならないよう、ピペットでの計量を正確にするよう
心がけましょう。
実験
Miniprep で調製したプラスミド DNA を使う。前の段階の塩基配列チェックで、期待
通りに変異の入っていた cDNA を使う。また、予想外の変異が入った cDNA が
あればそれも使う。約半数の人には cDNA の切断を担当してもらい、残り半数の
人には、受け入れ側(?)の pQE30 の切断を担当してもらう。
1. 1.5 mL エッペンチューブを用意し、cDNA 切り出し用の反応液を作る。
H2O 33 μL
Miniprep で調製したプラスミド DNA 5 μL
10x 制限酵素バッファー (a) 5 μL
10x Bovine Serum Albumin (b) 5 μL
BamHI (c) 1 μL
HindIII (d) 1 μL
2. 1.5 mL エッペンチューブを用意し、pQE30 切断用の反応液を作る。
注: “pQE30” とは言うものの何も組み込まれていないままのベクターではなく、
変異の入っていない EGFP cDNA が組み込まれたものを使う。それには
いくつかの理由があるので、後でみんなで考えてみよう。
H2O 34 μL
pQE30 (0.25 μg/μL) 4 μL
10x 制限酵素バッファー (a) 5 μL
10x Bovine Serum Albumin (b) 5 μL
BamHI (c) 1 μL
HindIII (d) 1 μL
3. 反応液をよく混ぜて、37℃ で約 1 時間、反応させる。
注: 制限酵素は、タンパク質なので、凍結融解を繰り返すと立体構造が
壊れて活性が落ちる。そこで、-30℃ の冷凍庫で凍らないように(また、
タンパクが安定に保たれるように) 50% のグリセリンを含むバッファー
に溶けている。そのため、反応液に酵素を入れると酵素はすぐに反応
液に混ざらずに底に沈む。反応液を作ったときによく混ぜないと、反応
が進まないので注意。
4. 反応液に等量のフェノール/クロロホルム混合液を加え、ボルテックス。
5. 5 分間遠心する。
6. 黄色の下層(有機溶媒の層)と無色の上層(水層)に分離するので、上層を
とり、新しいエッペンに移す。
7. 5 μL の 3 M NaOAc (e) と 125 μL のエタノールを加え、よく混ぜる。
注: これがごく標準的なエタノール沈殿(略してエタ沈)。
8. -80℃ に 5 分ほど置いた後、10 分間遠心し、上清を完全に捨てる。
9. 75% エタノールを 200 μL ほど静かに注ぎ込み、再び 1 分ほど遠心。
10. エタノールを完全に捨てる。11. 沈殿が乾燥したら 10 μL の TE を加える。
ここまでできたら次のステップへ。。。
実験に使う試薬・器具
メーカーによって多少の違いがあるが、例えばタカラが BamHI 用に奨める 10x(b) Bovine Serum Albumin
バッファーの組成は [200 mM Tris-HCl (pH 8.5)、100 mM MgCl2、10 mM DTT、
1000 mM KCl]。HindIII 用は [100 mM Tris-HCl (pH 7.5)、100 mM MgCl2、10 mM
DTT、500 mM NaCl]。両者の組成は異なるが、HindIII は BamHI 用のバッファーで
もよく切れる。一方、BamHI は HindIII 用のバッファーでは、20% 以下の相対活性
しか望めない。従って、今回の実習では BamHI 用のバッファーを使って、
両方の酵素で同時に切断する。
1 mg/mL のウシ血清アルブミンを使う。反応液中の制限酵素は低濃度になるが、(c) BamHI
ウシ血清アルブミンを混ぜておくと、タンパクの安定性が増す。酵素によっては
添加不要なのだが、ウシ血清アルブミンを添加することによって活性が阻害される
ような制限酵素はないので、(必要か不要かをいちいち考えるのが面倒なので)
大抵はウシ血清アルブミンを含む反応液を作る。
Bacillus amyloliquefaciens H という細菌起源。認識配列は [5'-G/GATCC-3']。(d) HindIII
Bam というのは属名の頭 1 文字と種名の頭 2 文字をとったもの。このように、
制限酵素の名前の頭 3 文字は学名(ラテン語)に由来するので、イタリックで書く。
4 文字め以降はイタリックにしない。ちなみに、DNA 上の認識配列を表記するときには
BamHI はイタリックにはしない。 BamHI は、高濃度のグリセリン、Mn2+ の存在、
低イオン強度下で Star 活性が出ることが知られている。
Haemophilus influenzae Rd という細菌由来。認識配列は [5'-A/AGCTT-3']。(e) 3 M NaOAc
Mn2+ や DMSO の存在下で Star 活性が出ることがある。
3 M の酢酸ナトリウムをこう書く。酢酸ナトリウムの粉を最終濃度 3 M になるように
溶かし、酢酸で pH を 5.2 に合わせたもの。核酸のエタノール沈殿用の塩としては
最も一般的に使われる。エタノール沈殿のときは 0.3 M になるように核酸溶液に
加え、さらに 2〜2.5 倍量のエタノールを加えて核酸を沈殿させる。(RNA の場合は
3 倍量のエタノールを使用する)