4. プラスミド DNA の塩基配列確認
はじめに
皆さんは、Kunkel 法によって突然変異を導入した “つもり” のプラスミドを大腸菌に導入し
コロニーを拾ってプラスミドを精製しました。このプラスミド中の EGFP cDNA には突然変異が
入っている “はず” です。ところが、ものごとはそれほど単純ではありません。さまざまな
原因で、期待した箇所に突然変異が入っていないプラスミドや、予想外の部分に突然変異の
入ってしまったプラスミドが回収されてくることがあります。したがって、期待したとおりの箇所
に期待したとおりの配列の突然変異が入っているかどうかを確認する必要があります。
比較的長い欠失変異や挿入変異を導入する場合には、PCR
で変異導入箇所を含む
cDNA 断片を増幅して電気泳動をすることである程度目星をつけることはできます。また
点突然変異の導入によって、その箇所に新たに制限酵素の認識配列ができたり、逆に
そこにあった制限酵素認識配列がなくなったりした(そのようにプライマーを設定した)
場合には、制限酵素の切断パターンを、やはり電気泳動で見ることによって目星をつける
ことができます。しかし、いずれにしても最終的にはきちんと塩基配列を確認しなければ
タンパクを発現させる実験に使うことはできません。
知っておかなければならないこと
塩基配列の決定法として現在広く使われている方法は Sanger et al.
(1977) が発明した
ジデオキシ法です。この方法の原理については他の講義で何度も聴かされることと思います。
ここでは原理については簡単に触れるだけにして、より実際的な方法を説明します。
塩基配列決定用の試薬や機器はいろいろなメーカーから市販されているので、実際に行う
作業はそれぞれのメーカーの説明書のとおりに進めれば間違いありません。そんなことも
あって、方法には実にさまざまなバリエーションがあります。ここでは、最初できるだけ一般的
な話をして、徐々にこの実習で実際に行う方法に関する説明を混ぜていきます。
ジデオキシ法では、塩基配列を決定したい DNA を鋳型として、それに相補的な鎖を合成
する反応を利用します。鋳型とする DNA は 1 本鎖でもいいし 2 本鎖の DNA 断片(例えば
PCR 産物)でもいいし、プラスミドそのままでも OK です。 1 本鎖を精製した方がきれいに
配列を読むことができるからということで pBluescript などの場合には f1 origin を利用して
調製した 1 本鎖 DNA を鋳型とする人がいますが、今回の実験では 2 本鎖のプラスミドを
そのまま鋳型として使います。最近のシステムでは、2 本鎖 DNA の配列でもとてもきれいに
読むことができます。
塩基配列のための DNA 合成反応では、基質となる dNTP に一定の割合でジデオキシ
リボヌクレオシド三リン酸(ddNTP)を混ぜて合成反応を行います。
ddNTP には 3' 末端に
OH 基がないため、鎖の伸長がそこで止まってしまうという性質を利用しています(図
1)。
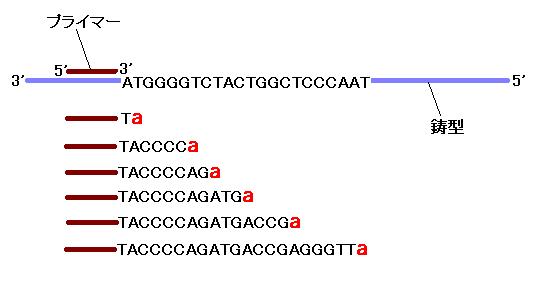
図 1
図 1 には反応液中に 4 種類の dNTP と ddATP を混ぜたときの反応 生成物を描いてあり
ます。図では ddATP が取り込まれた部分を a
で表し、正常な dATP が取り込まれた箇所
(A で表してある)と区別しています。どの程度の割合で ddNTP を混ぜるかは使う酵素に
よって異なります。 ddNTP の割合が高すぎると、プライマーに近い位置で全ての鎖の伸長
が止まってしまい、プライマーから遠い部分の配列が読めないなどの不都合が生じます。
この ”割合” は、使う DNA 合成酵素が ddNTP を ”どの程度の頻度で” 正常な dNTP と
間違うかによって決まります。
さて、このようにしてさまざまな箇所で合成が止まった、さまざまな長さの鎖を ”どうやって
見るか” です。今回の実験では ddNTP が蛍光色素で標識されています(蛍光色素が
ddNTP に共有結合でくっついています)。新たに合成されて ddNTP で伸長が止まった鎖が
蛍光色素で標識されていることになるわけです。これをポリアクリルアミドゲル電気泳動に
よって分離します。泳動しているゲルにレーザーを照射して蛍光を検出し、あとはコンピュー
ターで自動解析をするわけです。昔は蛍光色素ではなく放射性同位元素で標識し、泳動後
のゲルを X 線フィルムに押しあてて感光させ、それを暗室で現像していました。
さて、昔話はそれくらいにして、図 1 のような反応を ddCTP や ddGTP、ddTTP などに
ついてそれぞれ別々に行って、それら 4 サンプルを並べて電気泳動すれば、図 2 のような
泳動像を得られるはずです。
図 2 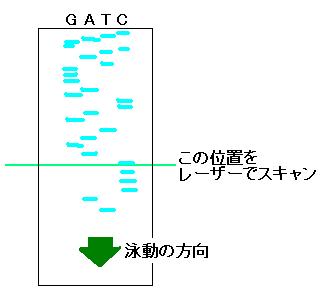
実際には、同じ鋳型から別々に合成反応を行って調製したサンプルを 4 レーン泳動する
というやり方はほとんど誰もやっていません。 1 つの反応液に 4 種類すべての ddNTP を
入れて 1 つのサンプルを 1 レーンだけ泳動するのです。どうしてそういうことができるのか
わかりますか?実は、各 ddNTP に異なる波長(色)の蛍光を発する別々の色素が結合
しているものを使うのです。ですから、全てのバンドが 1 レーンに泳動されていても、その
バンドの発する蛍光の色を検出することにより塩基配列を読むことができるのです(図 3)。
図 3 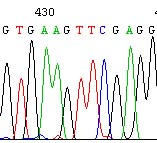
今回用いるのは Applied Biosystems 社の Big Dye シークエンス試薬キットを用いて
反応を行い、同社の 3100-Avant Genetic Analyzer
を用いて電気泳動と解析を行う
予定です。このシステムは、従来のポリアクリルアミドゲルを使いません。同社製の特殊
ポリマーを充填した細い管(キャピラリー)の中をサンプルが電気泳動されます。大きな
ゲル(昔は長いものでは縦が 1 m 近くあるものもありました)を作る手間がいらず、多数の
サンプルを自動的に処理できるという点でとても便利になっています。
実験
前の実験で調製したプラスミド DNA を使う。1. 0.5 mL の PCR 用チューブを用意し、以下の反応液を作る。
注: 4 人分を作る。通常、4 本の反応を行いたいときには 5 本分作って
4 本分を使って残りを捨てるのだが、今回は試薬がもったいないので
4 本分ちょうどを調製する。・・・ピペットでの計量を正確に!
H2O 25 μL
Reaction Mix (a) 2 μL
5x Sequencing Buffer (b) 7 μL
1.6 pmol/μL プライマー (c) 4 μL
2. 3 本の新しい PCR 用チューブを用意し、計 4 本のチューブに均等に
(9.5 μL ずつ)反応液を分注する。
1 人 1 本の反応液を使う。
3. 分注した反応液に、自分の miniprep DNA を 0.5 μL 加えて混ぜる。
4. サーマルサイクラーを利用して、以下のプログラムで反応を行う。
96℃ で 1 分
[96℃ で 10 秒、50℃ で 5 秒、60℃ で 4 分] を 25 回繰り返す
4℃ でキープ
5. 10 μL の H2O、1.25 μL の 0.5 M EDTA (d)、60 μL のエタノールを加え
るよく混ぜる。
6. 室温に 15 分静置したあと 15 分間遠心する。
7. 沈殿が見えることを確認してから上清を捨て、100 μL の 75% エタノールを
加える。
注: エタノールを加えるときには沈殿に向かって強く吹き付けないこと。沈殿が
チューブからはがれると扱いにくくなる。
8. 軽く遠心した後、沈殿を吸い取らないように上清を完全に捨てる。9. 埃よけのラップをかぶせて、10 分程度乾燥させる。
10. 沈殿が乾燥したら(透明になって見えなくなるはず)、15 μL の Loading
Buffer (e) を加えてサンプルを溶かす。
11. 95℃ で 2 分間加熱して 2 本鎖を解離させた後、すばやく氷で冷やす。
12. 96 穴プレートに移し、電気泳動する。
機械の操作についてはその場で、機械の説明書を見ながら説明する。
13. 塩基配列の解析結果から、期待した突然変異が導入されているかどうかを
確認する。
解析結果の見方については実物を見ながら説明する。
実験に使う試薬・器具
組成は不明。酵素とバッファー、塩、そして 4 種類の蛍光標識のついた ddNTP(b) 5x Sequencing Buffer
が入っているはず。メーカーの説明書では、20 μL の反応液に対して 8 μL
入れるようになっているが、多くの研究室ではその 8 分の 1 の濃度で使う。
今回の実習でも 10 μL の反応液に 0.5 μL しか入れない。その分反応液の
バッファーや塩の濃度が下がって酵素反応の条件が悪くなる。これを補うのが
5x Sequencing Buffer の役割である。
これも組成は不明(企業秘密)。Reaction Mix を節約するために塩やバッファー(c) シークエンス用プライマー
成分を補うためのもの。メーカーがつけてくるということを考えても、ほとんどの
人が節約をしていることがわかる。
PCR と違って 1 つだけ使う。 2 つのプライマーを使ったらどうしてまずいのか(d) 0.5 M EDTA
考えてみよう。今回の実習では T3 プロモーターの配列のプライマーを使う。
プライマーがプラスミドのどの部分にアニーリングしてどちら向きの配列を
読むことができるか考えておくこと。
EDTA はエチレンジアミン四酢酸(ethylene diamine tetraacetic acid)。(e) Loading Buffer
二ナトリウム塩の粉末をミリ Q 水に溶かす。そのままでは溶けないので、NaOH
を加える。最初のうちは粒をそのまま入れていく。 pH を測りながら慎重に。
EDTA の粉がかなり溶けてきたら、粒ではなく 10 N の NaOH を少量ずつ加え
慎重に EDTA を溶かす。溶け切った時点で pH はかなり 8 に近いはず。
基本的にはホルムアミド。メーカー特製のものをそのまま使っている。
ホルムアミドは、核酸を変性させ塩基対を作りにくくする。 1 本鎖 DNA を泳動
するので、パリンドローム(回文構造)になっている部分で分子内塩基対合
が起こったりするのをホルムアミドが防ぐ。