海洋生命・分子工学実験 II
(8) リコンビナントタンパクの精製
はじめに
この実験で作ったプラスミド(pQE30)は,挿入 cDNA
がコードするタンパクを簡単に大量に発現
させるために巧妙にデザインされたものであることが,前の章までの実験でわかったと思います。
しかし,このプラスミドの便利さはまだまだ他にもあるのです。日程の都合上,実際は作業でき
ないかも知れませんが,ここで紹介するのは大腸菌に発現させたリコンビナントタンパクを
ワンステップで簡単に精製する手法です。このような方法で合成されるリコンビナントタンパクの
量は,確かに大腸菌が本来持っているどのタンパクよりも多いくらいです。しかし,それでも
SDS-PAGE の結果からもわかるように,皆さんがほしいタンパクは,(それぞれの量は少ない
とはいえ)数え切れない大腸菌タンパクの中に混ざって存在しているというのも事実です。例えば
皆さんはリコンビナントタンパクの機能を詳細に解析したいとします。そのとき,もし大腸菌の中に
そのタンパクとよく似た・・・しかし何十倍・何百倍の強い活性をもつタンパクがあったとしたら,
例え量が少なくてもそのタンパクの存在は無視できなくなり,実験結果も信用できないものになり
ます。そこで,できることなら目的のタンパクだけを精製しなければなりません。今回使った
pQE30 のようなプラスミドは,作ったタンパクを精製するために便利な工夫がほどこされています。
知っておかなければならないこと
もう一度 pQE30 の multi-cloning site の配列(図 1)を見てください。今回注目してほしいのは
pQE30 によってコードされている N 末端近くに 6 個連続して存在するヒスチジン(H)です。
図 1 
この 6x ヒスチジンリピートが,リコンビナントタンパクを精製するための便利な道具なのです。
図 2
を見てください。右側にうぐいす色の壁があります。これは固相をあらわしています。
実習で使うとすれば,この部分はアガロースゲルです。固相から飛び出している枝の先(左端)
の部分を
Nitrilotriacetic acid (NTA) といいます。NTA
は,ニッケルイオン(Ni2+)の 6 つの
結合箇所の 4
ヶ所を捕まえています。つまり 2 ヶ所がフリーです。その残った 2
ヶ所に
ヒスチジンが結合するのです。図では左側の青いジグザグの太線がタンパク質のペプチド
骨格をあらわしていて,そこから 2
個のヒスチジン側鎖が出ている様子を示してあります。
図 2 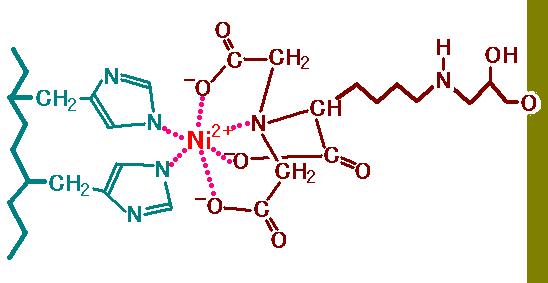
この性質を利用したアフィニティークロマトグラフィーによって,リコンビナントタンパクを簡単に
精製できるのです。N
末端に付加されたヒスチジンリピートは,たくさんの荷物の中から自分の
荷物を見分けて選び出す荷札(タグ)にたとえて,ヒスチジンタグ (His-タグ)と呼ばれます。
His-タグはタンパク全体の立体構造を壊すほど大きくないので,多くの場合タグをつけたまま
活性の測定などの実験に使うことができます。もしも,タグがそのタンパクの機能をじゃまする
ような場合には,タグを
C 末端につけることもできます。この実験に使った pQE シリーズの他
にも,タンパク発現・精製用のプラスミドはたくさんあります。例えば、Amersham から売られて
いる
pGEX ベクターは、約 26 kDa のグルタチオン-S-トランスフェラーゼ(GST)の翻訳
領域の下流に自分の cDNA を組み込んだ融合タンパクとして発現させるシステムです。
この場合,融合タンパクは GST がグルタチオンと結合する性質を利用したアフィニティー
クロマトグラフィーによって精製することができます。精製したタンパクは融合タンパクのまま
活性測定等に使うこともできますし GST 部分と切り離すこともできます。GST
翻訳領域の
下流、cDNA との連結部のすぐ上流にトロンビンというプロテアーゼの認識配列 [ LVGRPS ]
があって,そこで切り離すことができます。また,New England Biolabs で売っている pMAL
ベクターは約 42 kDa のマルトース結合タンパク(MBP)との融合タンパクを作れます。この
場合は MBP
がマルトース(麦芽糖)と結合することを利用したアフィニティークロマトグラフィー
で精製できます。また精製後に MBP の下流にある Factor
Xa (これも血液凝固系で働く
プロテアーゼです)認識部位 [ IEGR ] を切断することによって,MBP
部分と切り離すことが
できます。
実験
1. 前の章と同様に IPTG
でタンパク発現を誘導した大腸菌をエッペンに移し
遠心して集菌する。菌の沈殿を残し上清を捨てる。
グループで 1 サンプル。前の晩からの大腸菌の培養と IPTG によるタンパク発現
誘導は藤原がやっておきます。
2. エッペンに バッファー B
(a) を 1
mL 注ぎ,ボルテックスで混ぜる。
3. 10 mL 用チューブに移し,超音波破砕機で合計 3 分ほど
sonication。
1 回に 1 分程度。氷冷しながら行う。30 秒ほど休んで再び超音波・・・を繰り返す。
大腸菌が破壊されれば液が透明になる。
4. 溶菌液を新しいエッペンに移し, 20〜30 分間,冷却遠心する。
5. 遠心の間に Ni2+‐NTA‐アガロースビーズをバッファー B
で平衡化する。
(1) 50 μL のビーズをエッペンにとり,まずビーズを懸濁してある液を捨てる。
(2) 500 μL のバッファー B を加える。
(3) マイクロピペッタ−で混ぜた後,低速で遠心し,上清を捨てる。
(4) 再びバッファー B を 500 μL 加えて混ぜる。
(5) 低速で遠心し,上清を捨てる。(上清を捨てるのは,溶菌液の遠心が終わる
直前でよい)
6. 溶菌液の遠心が終わったら,上清のうち 50 μL を別のエッペンにとり,等量の
2x
SDS-サンプルバッファーと混ぜる。残りの上清を平衡化済みのビーズの
入ったエッペンに移す。
液はたくさん残してもいいので,沈殿を絶対にとらないように。
7. 30 分ほど室温に置き,ときどきピペッタ−で混ぜる。
Ni2+‐NTA‐ビーズに His-タグ付きリコンビナントタンパクを吸着させる。
8. 低速の遠心でビーズを沈殿させた後,上清を捨てる。
9. ビーズに バッファー C (b)
を 1 mL 加えて混ぜる。
バッファー C はビーズの洗浄用。このバッファー中では His‐タグタンパクはビーズ
から離れない。ビーズの隙間などに(Ni2+-NTA に吸着したわけでもないのに)
残っている不要なタンパクを除去する。
10. 手回し遠心でビーズを沈殿させ,上清の 50 μL を別のエッペンに移し,
等量の 2x SDS‐サンプルバッファーを加える。残りの上清を捨てる。
11. ステップ 9 と 10 をもう 1 度繰り返す。
ただし SDS-PAGE 用のサンプリングはしなくてよい。
12. ビーズに バッファー E'
(c) を 100 μL 加えて混ぜる。
バッファー E' には
イミダゾール(d) が入っている。これによって His-タグタンパク
と Ni2+-NTA との結合が阻害され,タンパクはビーズから離れる。
13. 室温で 5 分ほど置き,ときどき混ぜる。
14. 低速の遠心でビーズを沈殿させ,上清を新しいエッペンにとる。このうち
50 μL を別のエッペンにとり,等量の 2x SDS‐サンプルバッファーを
加える。
15. ステップ 12〜14 をもう 1
度繰り返す。
ただし SDS-PAGE 用のサンプリングはしなくてよい。
16. SDS‐サンプルバッファーに溶かした各サンプルを 80〜90℃ で 5 分煮て
15〜20 μL 程度を電気泳動する。
実験に使う試薬
(a) バッファー B
8 M 尿素
100 mM リン酸二水素ナトリウム(NaH2PO4)
10
mM Tris-HCl (pH 8.0)
(b) バッファー C
8 M 尿素
100 mM リン酸二水素ナトリウム(NaH2PO4)
10
mM Tris-HCl (pH 6.3)
(c) バッファー E’
8 M 尿素
100 mM リン酸二水素ナトリウム(NaH2PO4)
10
mM Tris-HCl (pH 8.0)
250 mM イミダゾール (下を参照)
(d) イミダゾール
図 3 の左がヒスチジン,右がイミダゾール。図 2 と見比べよう。イミダゾールが大量に
持ちこまれると,このイミダゾールが
Ni2+-NTA と結合して,その結果 His‐タグタンパクが
ビーズから離れる。
図 3 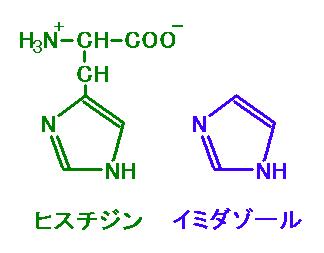
遺伝子工学的実験法の目次に戻る