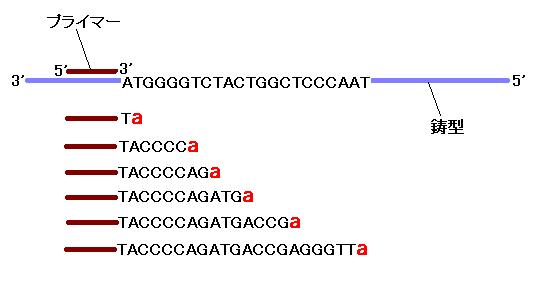
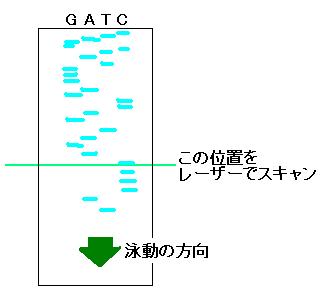
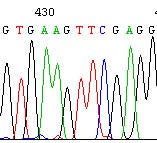
(4)
の実験で精製したプラスミド DNA を鋳型として使います。
1.
0.5 mL の PCR 用チューブを用意し,以下の反応液を作る。
注: 10 サンプル分を作ります。通常 10 本の反応を行いたいときには 11〜12 本分作って
余りを捨てるのですが,今回は試薬がもったいないので 10 本分ちょうどを調製します。
ピペットでの計量を正確に!
2.
9 本の新しい PCR 用チューブを用意し,計 10 本のチューブに均等に(9.5 μL
ずつ)反
応液を分注する。
1 グループ(3〜4 人)で 1 本(全部で 10 本)の反応液を使います。
3.
分注した反応液に,プラスミド(miniprep 産物)を 0.5 μL 加えて混ぜる。
4.
サーマルサイクラーを利用して、以下のプログラムで反応を行う。
96℃ で 1 分
[96℃ で 10 秒,50℃ で 5 秒,60℃ で 4 分] を
25 回繰り返す
4℃ でキープ
反応終了後は,藤原が片付けます。以下の操作は次の実習の日に行います。
5.
10 μL の H2O,1.25 μL の 0.5 M EDTA (d),60 μL のエタノールを加えて
混ぜる。
6.
室温に 15 分静置したあと 15 分間遠心する。
7.
沈殿が見えることを確認してから上清を捨て,100 μL の 75% エタノールを
加える。
注: エタノールを加えるときには沈殿に向かって強く吹き付けないこと。沈殿が
チューブからはがれると扱いにくくなります。
8.
軽く遠心した後,沈殿を吸い取らないように上清を完全に捨てる。
9.
埃よけのラップをかぶせて,10 分程度乾燥させる。
10.
沈殿が乾燥したら(透明になって見えなくなるはず),15 μL の Loading
Buffer (e) を加えてサンプルを溶かす。
11.
95℃ で 2 分間加熱して 2 本鎖を解離させた後,すばやく氷で冷やす。
高知大学遺伝子実験施設のシークエンス解析装置で解析していただいている
ので,ここから先は自分たちではやらず,遺伝子実験施設に解析していただき
ます。
12.
96 穴プレートに移し,電気泳動する。
13.
塩基配列の解析結果から,期待したとおりに組み立てられているかどうかを
確認す
る。
解析結果の見方については実物を見ながら説明する予定。