海洋生命・分子工学実験 II
この実習では,ここまで pBluescript II SK+ というプラスミドを使って,mCherry タンパク質を
コードする cDNA をクローニングしてきました。 しかし,実は,pBluescript II SK+ は,もともと
大腸菌を使って外来遺伝子のコードするタンパク質を発現させる目的には向いていないの
です。そこで,この実験だけは,別のプラスミドに組み込まれた mCherry を発現させます。
そのプラスミドは pQE31
(QIAGEN 社) といいます。
pQE31 の配列
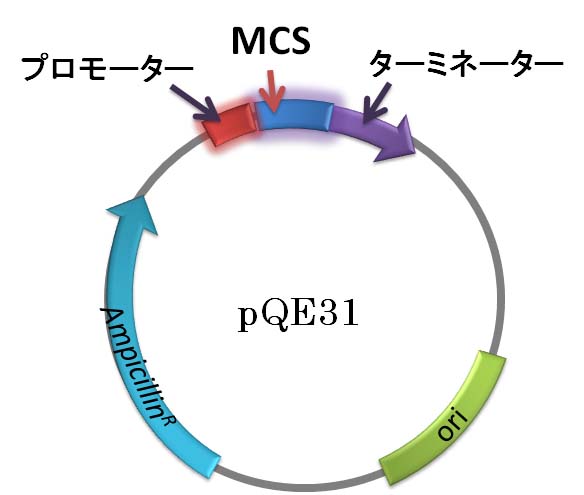
pQE31 は,全長 3463 bp のプラスミドです。複製起点 (ori) やアンピシリン耐性遺伝子
(ampr) をもつところは pBluescript II SK+ と共通です。しかし,pQE31 には,それらに加えて
組み込んだ外来 DNA がコードするタンパク質の発現をコントロールする仕掛けがあります。
その部分の配列を下図に示します。上の図ではプロモーター (promoter) と MCS (マルチ
クローニングサイト) にあたる部分です。
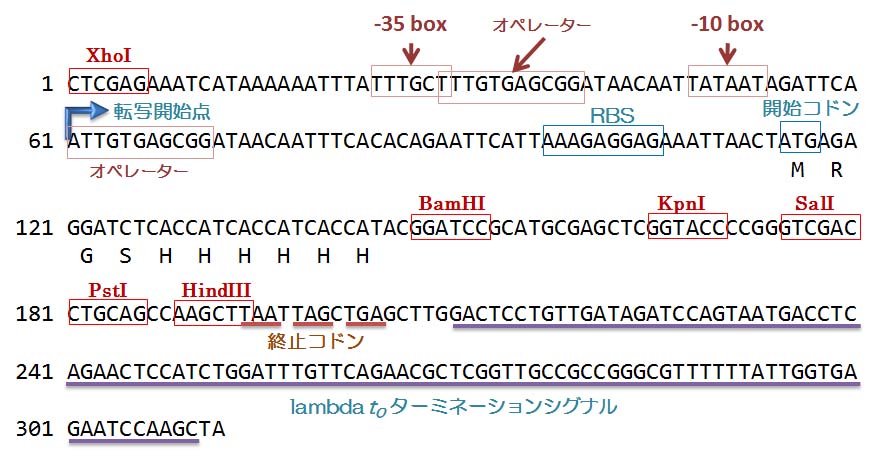
制限酵素 XhoI の認識配列 (CTCGAG) の直後に,バクテリオファージ T5
のプロモーター
配列があります。このプロモーターは非常に強力です。ですから,ひとたびこのプロモーター
から遺伝子が発現すると,そこから生産されるタンパク質の量は,多くの場合大腸菌自身が
もともと持つどのタンパク質よりも多くなります。 プロモーター配列とオーバーラップして,
lac
オペレーターの配列が見られます。転写開始点から
37 塩基下流に RBS (AAAGAGGAG)
がありますね。そしてその直後に開始コドンがあります。そのちょっと下流に,制限酵素
BamHI
の認識配列があります。そこから,制限酵素 HindIII
の認識配列までの間には,
たくさんの制限酵素認識配列があります。ここが pQE31 の multi-cloning site (MCS)であり,
この制限酵素を利用して自分の興味のある cDNA などを組み込むのです。最後の HindIII 配列
の直後から終止コドンが 3 つ並んでいます。自分の組み込んだ cDNA が終止コドンを含んで
いなくても,ここで必ず翻訳が終了するようにできています。それに続いて転写終結のシグナル
であるターミネーター配列がありますね。
さて,pBluescript II SK+ と大きく異なるのは,組み込んだ外来 DNA を転写するための
プロモーターがきわめて強力であることです。タンパク質をたくさん作りたいから,そうする
のですね。。。ところが,このプロモーターはあまりにも強力なため,大腸菌自身のラクトース
リプレッサーでは抑制しきれず,だらだらと発現してしまうことになります。私たちが作りたい
(研究したい) タンパク質というのは,生物の細胞の中で何らかの重要な役割を演じる (ことが
期待される) タンパク質ですよね? そうだからこそ,「研究したい」 と思うわけです。でも
そういうタンパク質は,大腸菌の細胞の中でも何かしら仕事をしようとして・・・その結果として
大腸菌の生育にとって障害になることが多いのです。実際,T5 プロモーターからの転写が
抑制されないような状態でプラスミドを大腸菌に導入し,増殖してコロニーをつくった大腸菌
を回収すると,その中には正常はプラスミドが入っておらず,プロモーターに突然変異が
できてしまったようなプラスミドばかりが取れてきたりします。・・・つまり,正常に遺伝子を
発現できるようなプラスミドが入った大腸菌は増殖できなかったというわけです。。。
そこで,以下のようなことが大切になってきます。
(1) pBluescript II SK+ は,大腸菌の細胞 1 個あたりに数百個程度にまで増殖します。
これに対して,pQE31 は大腸菌の細胞 1 個あたり数個にしかなりません。プラスミドの
コピー数が少ないのです。このことは,プラスミド DNA 自体を大腸菌から精製したり
その配列を決定したりするときには厄介な特徴なのですが,タンパク質を発現させよう
という場合には,その方が好都合なのです。
(2) pQE31 などのプラスミドを導入する大腸菌の系統自体も,特別なものを使います。
何種類もあるのですが,多くに共通してみられる特徴の一つとして,強力なリプレッサー
をもつことがあげられます。ラクトースリプレッサーをコードする遺伝子 lacI に突然変異
をもつ lacIq という遺伝子が使われます。この突然変異型のリプレッサーは,野生型の
リプレッサーよりずっと強力なので,T5 プロモーターからの転写を抑制することができます。
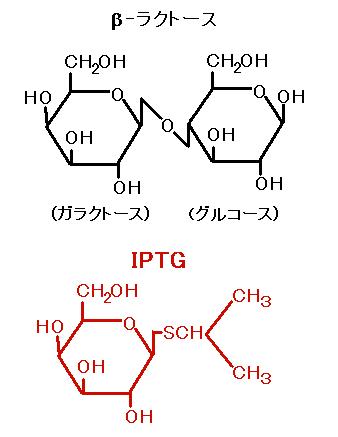
これらのことにより,pQE31 を導入された大腸菌では,ラクトース (実際にはラクトースと
構造の似たイソプロピルチオ-β-D-ガラクトシド = IPTG) で誘導をかけるまで,遺伝子の
発現を抑制した状態で大腸菌を増殖させることができるわけです。そうして,ある程度
大腸菌を増殖させたところで,“いまだ!” という具合に IPTG を添加して遺伝子発現を
誘導し,大量のタンパク質を得ることができるのです。
さて,この実験で使うプラスミドは,下記の手順で作りました。
(1) pCRII-mCherry (← 何のこと?なんて人はいないだろうね) を BamHI と HindIII で切りました。
(2) その切断片をアガロースゲル電気泳動で分離し,mCherry cDNA をゲルから切り出しました。
(3) 同様に pQE31 を BamHI と HindIII で切り,プラスミド本体をアガロースゲルから切り出しました。
(4) (2) と (3) の断片を混ぜ,ライゲーションを行いました。
(5) ライゲーション反応液を用いて,大腸菌 JM109 系統と RB791 系統のトランスフォーメーション
を行いました。どちらの大腸菌も上述の lacIq をもつ系統です。
(6) 得られたコロニーを,コロニー PCR で検査しました。
(7) コロニー PCR に合格したクローンの塩基配列を決定し,pQE31 に正しく mCherry cDNA が
組み込まれていることを確認しました。
さて,みんな,このプラスミドがどんな配列をしているか,もうこれ以上説明する必要はないね?
実験
みんなに,大腸菌がほど良く増殖中の試験管 (0.5 ml の LB/amp 培養液中に菌がいます) を
渡します。
1. 約半分の人は,試験管の大腸菌に, 2 mM IPTG
を含む
LB/amp 培養液を 0.5 ml 加える。
残りの人は,IPTG を含まない LB/amp 培養液を 0.5 ml 加えて,室温で振盪培養する。
IPTG を入れたときと入れなかったときの SDS-PAGE の泳動パターンを比較します。
2. 室温で実習終了直前まで培養した後,試験管の大腸菌をエッペンチューブに移し
遠心して菌を沈殿させる。
沈殿した大腸菌を見てみましょう。タンパクが発現していれば赤いはずです。 IPTG を含む
培養液と,含まない培養液の大腸菌で,色の違いはありますか?
3. 上清を捨て,SDS サンプルバッファーを加えてボルテックスで混ぜる。
SDS サンプルバッファーの組成については,専門海洋生命・分子工学基礎実験
(藤原
担当)あるいは海洋生命学実習 I
(湯浅先生担当)のレジュメを参照。
4. 80℃ から 90℃ 程度で 5 分煮て,菌を溶かす。
注: 菌が溶けると染色体 DNA が出てきて液がネバネバするので,ボルテックスをして
DNA を断片化し,液の粘性を下げておきます。この日の作業はここまでです。
次の時間に SDS-PAGE を行います。
5. SDS-PAGE によってタンパクを分離し,クマシーブルー染色をする。
SDS-PAGE に関する資料は別に配ります。
遺伝子工学的実験法の目次へ戻る