7. DNA 断片の連結(ライゲーション)
はじめに
ゲルから精製した DNA 断片(変異の入った EGFP cDNA 断片と、pQE30 プラスミド)を
連結して環状プラスミドを完成させます。どのようなプラスミドになるかは第 0 章( こちら )
を見て考えてください。
知っておかなければならないこと
cDNA とプラスミドの連結に使うのは T4 ファージ由来の T4 DNA ligase という酵素です。
この酵素が触媒する反応は、末端の形状が同じ DNA どうしの塩基対形成ではありません。
DNA (または RNA)鎖の 5' リン酸基と、3' 水酸基を結合させる・・・すなわち、DNA の
ホスホジエステル結合を作る反応です(「分子遺伝学 C」の復習)。この実験では、
Promega という会社で売っている反応バッファーを使いますが、その組成が秘密になって
います。
そこで、キットを使わない場合の基本的な方法を簡単に紹介します。 T4 DNA ligaseは、
例えば以下のような組成の反応液で使用します。
[50 mM Tris-HCl (pH 7.5), 10 mM MgCl2,
10 mM DTT, 1 mM ATP]
MgCl2 や DTT、ATP は必須です。通常の方法では室温または
14-16℃ 程度で 4‐16
時間ほど反応させます。T4 ファージが感染した大腸菌の細胞内では、酵素は 37℃ 付近
で働きます。実際 37℃ で反応させた方が速いのですが、酵素の熱安定性が低いので
早く失活してしまいます。そこで、低温で反応させるわけです。
今回のように、2 種類の DNA 鎖を連結したい場合、DNA 鎖どうしが出会う確率が高い
ほど、ライゲーションの効率が高くなります。したがって、反応液にポリエチレングリコール
のような “場所をとる” ポリマー分子を高濃度で混ぜておくと、溶液の全容量の中で
DNA が動き回れる “スペースが非常に狭く” なり、ライゲーション効率が上がります。
キットでは、おそらくこのような工夫がなされており、そのため、室温程度でも非常に
効率よくライゲーションが起こります。
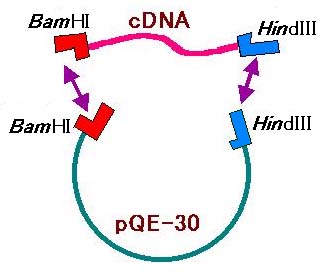
今回のライゲーションでは、プラスミドを BamHI
と HindIII で切断してあるので
プラスミド断片の両末端の形状が違います。したがって、“切れなかった” とか “片方の
酵素でしか切れなかった” というような “切れ残り” がない限り、1本のプラスミドの両端が
cDNA を挟まずに連結して閉じてしまうこと(セルフライゲーションという)はありません。
また cDNA どうしが結合することもありません。したがって、連結後に環状になったプラス
ミドは高率で cDNA を挟みこんでいることが期待されます。
1. 0.5 mL の PCR チューブに変異の入った EGFP cDNA と pQE30 を入れ
合計 4 μL にする。
注: ゲルから精製した DNA 断片を用意する。変異の入った cDNA を持った人と
pQE30 プラスミドを持った人が 1 人ずつペアになり、断片を交換して
ライゲーションの反応液を作る。このとき、各自の DNA 断片の、精製後の
電気泳動の結果を持ち寄り、cDNA とプラスミドのバンドの明るさがほぼ
等しくなるように量を決める(目安は、弱いけれどもはっきりとバンドが見える
程度)。その際、両方の断片の量の合計が 4 μL 以下になるようにする。
かすかにでもバンドが見えれば十分。多すぎるとうまく行かない。2. 以下の試薬を加えて反応液を作る。
2x ライゲーションバッファー (Promega) 5 μL
T4 DNA ligase (Promega) (a) 1 μL
3. 室温で 1 時間反応させる。
4. 第 1 章の「1 日め」( こちら )の方法で大腸菌のトランスフォーメーションを
行う。
注 1: ただし、RB791 (b) 系統の大腸菌の competent cells (50 μL)を使い、
クロラムフェニコールを含まない LB/amp 寒天培地にまく。
注 2: ライゲーション反応液は 5 μL を使い、残りは冷蔵保存しておく。
5. 一晩培養し、翌日、プレートのコロニーを確認し、次のステップへ。
実験に使う試薬・器具
2 本鎖 DNA の付着末端あるいは平滑末端の 5' リン酸基と 3' 水酸基を連結する(b) RB791
酵素。二重鎖 DNA どうしだけではなく、二重鎖 DNA と二重鎖 RNA、あるいは
二重鎖 RNA の末端どうしを連結する活性もある。ただし 1 本鎖の核酸を連結する
ことはできない。
ここで使う大腸菌は、タンパク発現用のものが望ましい。CJ236 や DH5α は
使わない。CJ236 は、ウラシル DNA グリコシラーゼ等の遺伝子が欠損しており
DNA 損傷に対する修復の能力が弱い(ということは変異を起こしやすい)。また、
DH5α など通常のプラスミドのクローニングに使う大腸菌のもつ lac リプレッサー
(野生型の lacI 遺伝子産物)では、pQE30 などの発現ベクターの持つ強力な
プロモーター(pQE30 の場合には T5 ファージの初期プロモーター)を抑制すること
ができない。・・・「実験でタンパクを発現させたい」と考えるくらいだから、そのタンパク
は何か生物学上重要な機能を持っているはずで、そのような機能性タンパクを大量
発現させたら、大腸菌の生育に害を及ぼす可能性がある。したがって、(例えば
次の章のような)実験のときには、まず大腸菌をある程度増殖させておいて、
それからタンパクの発現を誘導するという手順をとる。そこで、増殖中の大腸菌で
常時タンパクが発現することのないように、しっかりと発現を抑制しなければならない。
RB791 のような大腸菌は、変異を起こして強力になったリプレッサーをコードする
lacIq 遺伝子をもっており、pQE30 の強力なプロモーターを抑制できる。タンパク発現
用の大腸菌には、他にもさまざまな系統がある。中にはプロテアーゼの遺伝子を
欠損していて、外来タンパクを大量に発現させても内在性プロテアーゼによる分解を
受けないようになっている系統もある。