8. 変異タンパクの発現
《 みんなのデータ 》
はじめに
トランスフォーメーションの結果はどうでしたか?コロニーはたくさんできていましたか?
第 1 章の「知っておかなければならないこと」のところ(
ここ )に書いてあるように、
トランスフォーメーション後に増殖して LB/amp 寒天培地上にコロニーを作るのは
プラスミドをもつ細胞だけです。しかし、コロニーを拾ったときに、その大腸菌が正しい
(期待したとおりの変異をもつ EGFP cDNA が組み込まれた)プラスミドをもっているか
というと、現実はそんなに甘くありません。拾ってみたら、変異のない EGFP プラスミドを
含んでいる可能性もあります。(例えば pQE30 が BamHI か HindIII
のどちらか一方
でしか切れていなくて、セルフライゲーションしてしまった・・・などの原因で。)そこで、
期待通りのプラスミドが入っているコロニーを選ぶために、いくつかのチェックを行う必要
があります。同じことは第 3 章( こちら )でも議論しましたね)。
プラスミドが本物かどうかを確かめる方法はいろいろあります。第 3 章では、点突然
変異の有無を調べるために塩基配列を調べました。もしも、導入する変異が比較的
長い配列の欠失(deletion)や挿入(insertion)であったとしたら、変異導入箇所を
はさむような cDNA 断片を制限酵素で切り出してアガロースゲル電気泳動を行い、
バンドの長さである程度の判断をすることもできます。プラスミドを鋳型として(あるいは
拾ったコロニーの懸濁液の一部を強引に鋳型として) PCR を行い、これを電気泳動
してサイズを見ることもできますよね?また、点突然変異だったとしても、導入した変異
によって本来そこにあった制限酵素認識配列がなくなったり、逆に新たにできたりした
場合には、制限酵素による切断のパターンでチェックすることもできます。このあたりの
ことは第 4 章( こちら
)に書きました。今回の実験でも、時間に余裕があれば pQE30
に変異 cDNA を組み込んだ後のプラスミドの塩基配列を確認しましょう。そして、
1 人 1 クローンを選んで、タンパクを発現させてみましょう。発現させたタンパクは
SDS-PAGE で確認しましょう。また、せっかく EGFP を発現させるのだから、光らせて
みたいですね。


さて、そもそもなぜ pQE30 にオペレーター配列をつけたのでしょうか?
それは、組み込んだ cDNA からのタンパクの発現をコントロールするためです。 我々が
研究の対象にしようとするタンパクは、たいてい何らかの重要な生理活性を持っています。
細胞の機能に大きな影響を及ぼすそのようなタンパクは、往々にして大腸菌にとっては
迷惑な(大腸菌の生育の負担になったり害になるような)タンパクなわけです。そこで、常時
発現させておくと大腸菌が生育せず、結局タンパクを得られないという結果になってしまい
ます。実際、EGFP のような毒にも薬にもならないように思えるタンパクでも、これを発現し
続ける大腸菌は生育が著しく遅くなります。そこで、タンパクを発現させる実験の場合、
まず組み込んだ遺伝子の発現を抑制した状態で大腸菌をある程度増殖させ、その後に
遺伝子発現を誘導するという方法がとられます。
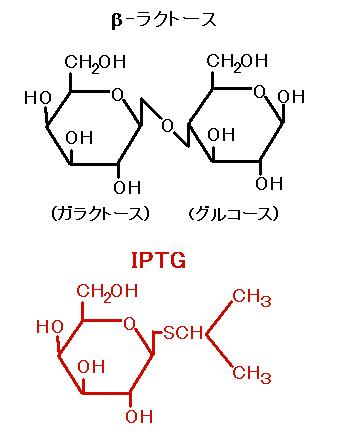
この実験では、ラクトースのかわりに、イソプロピル-β-D-ガラクトピラノシド
(IPTG)を
用いて転写を活性化します。IPTG(図 3)は、ラクトース同様にリプレッサーと結合し、
オペレーターから引き離しますが、ラクトースと違って分解されて減ることがないので、
長時間安定に転写活性を維持することができます。その結果、合成されるタンパクは
大腸菌の細胞の中で最も量の多いタンパクになります。
実験
前日〜1. トランスフォーメーション後のプレートから爪楊枝で大腸菌コロニーを拾い、当日〜
1.5 mL の LB/amp 培養液(試験管)に植えて一晩振盪培養する。
1 人 1 コロニーを、前の晩に教官が用意しておきます。皆さんはこの作業を
する必要はありません。
2. いっぱいに増えた大腸菌を約 100 μL とり、3 mL の LB/amp 培養液に
移して、さらに培養を続ける。
実習開始の数時間前に行う。
3. 培養液を 1 mL、エッペンに回収し、30 秒間遠心する。
4. 上清を捨て、50 μL の SDS-サンプルバッファーを加えてボルテックス。
これをタンパク誘導前のサンプルとする。
SDS サンプルバッファーの組成は物質科学実験 CIII のレジュメを参照。5. 残り 2 mL の培養液に 100 mM IPTG を 20 μL 加えて、37℃ でさらに
2 時間培養する。
6. 培養液を 500 μL、エッペンに回収し、30 秒間遠心する。
7. 上清を捨て、100 μL の SDS-サンプルバッファーを加えてボルテックス。
これをタンパク誘導後のサンプルとする。
8. 80℃ から 90℃ 程度で 5 分煮て、菌を溶かす。
注: 菌が溶けると染色体 DNA が出てきて液がネバネバするので、
ボルテックスをして DNA を断片化して液の粘性を下げておく。9. SDS-PAGE によってタンパクを分離し、クマシーブルー染色をする。
SDS-PAGE に関する資料は別に配ります。